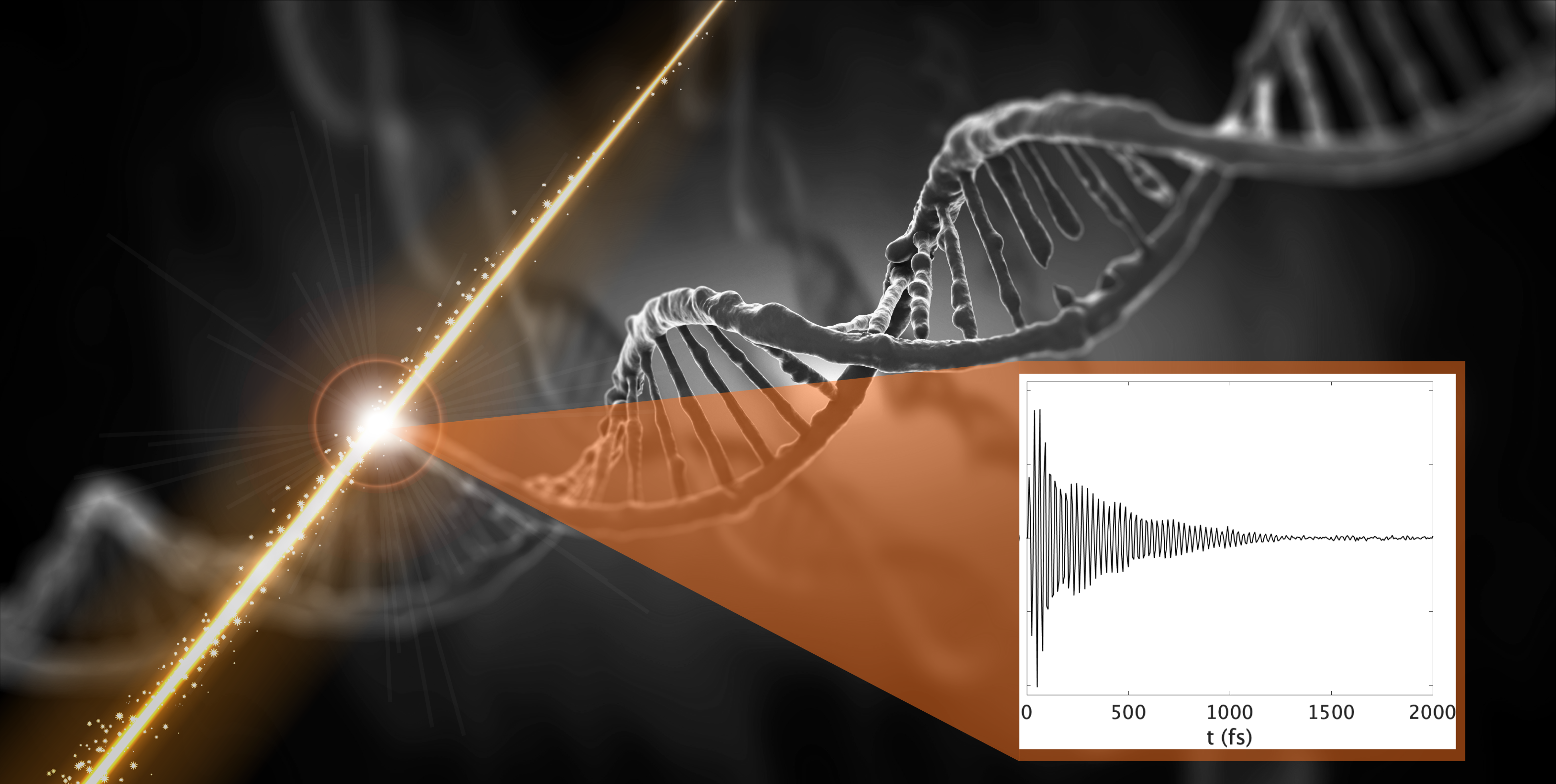
Quantum coherence promises more efficient—or sometimes entirely new—capabilities in areas like sensing, navigation, metrology, encryption, photovoltaics, and computation. However, rapid decoherence often stymies these goals.
This rapid decoherence happens due to environmental perturbations that destroy the phase relationship between participating states. By measuring, modeling, and understanding the molecular dependencies of this coherence, we are optimizing coherence lifetimes in chromophore networks.
We have developed tools using 2D electronic spectroscopy, computational modeling, and genetic algorithms to quantify these effects. Our next goal is to leverage high-throughput synthesis of systematically varying chromophore networks to establish machine-learning algorithms that can deduce subtle optimization trends from large sample sets. Our ultimate goal is to generate molecular optimization rules for prolonged electronic coherence.
A key lesson from biological pigment-protein complexes is that optoelectronic processes can be built into a system's dynamics by the precise arrangement of scaffolded, modular chromophores. Additionally, aggregation characteristics are especially important for powerful (but elusive) effects such as singlet fission, proton-coupled electronic transfer, and photoprotection.
Furthermore, with control over the aggregate structure of molecular networks, key ideas in fields like photovoltaics can be realized, such as having highly interdigitated donor-acceptor boundaries with consistent bicontinuous pathways to their respective electrodes.
These opportunities are being pursued using a combination of high-throughput automated synthesizers of self-assembling molecular networks, physical modeling, and optical experiments. In combination with the high throughput, they are bolstered using machine-learning methods to artificially evolve and optimize the systems.